


Modelagem Computacional da Matéria Condensada (MC)²
- Início
- DFMC - Departamento de Física da Matéria Condensada
- Modelagem Computacional da Matéria Condensada (MC)²
Selecione o seu idioma

")
")



O Grupo de Modelagem Computacional da Matéria Condensada utiliza simulações computacionais, juntamente com métodos analíticos, para investigar sistemas nanométricos (com tamanhos que vão de dezenas a milhares átomos), transições de fase em sistemas macroscópicos e defeitos em sólidos cristalinos. Estuda a formação de novas estruturas nanométricas, como clusters, nanofios, estruturas de carbono como nanotubos e grafeno. São abordados fenômenos ainda não bem compreendidos, como as transições líquido-líquido, as transições vítreas e a influência dos defeitos dos sólidos cristalinos nas propriedades gerais desses materiais.
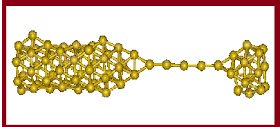
Modelagem computacional é uma das três abordagens existentes para pesquisa em Física, juntamente com a Física Teórica e a Física Experimental. Trata-se do uso intensivo de computadores para o estudo de fenômenos físicos. A Física Computacional funciona em íntimo contato com as abordagens teórica e experimental. Podemos, através da Física Computacional, testar modelos propostos pela Física Teórica, assim como investigar situações que estão além das possibilidades experimentais, como, por exemplo, materiais que estão em temperaturas e pressões extremas ou muito longe do equilíbrio.
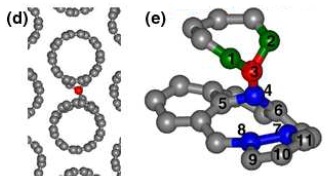
A modelagem computacional pode dizer, por exemplo, quais configurações os átomos assumem quando se coloca um defeito numa estrutura ordenada. Acima, o defeito é uma ligação entre dois nanotubos vizinhos. À esquerda, os nanotubos são vistos de cima, com a ligação em vermelho. À direita, detalhe do rearranjo dos átomos na vizinhança do defeito
Fonte: Silva et al., Nano Lett. 2005, 5 (6), 1045
O Grupo usa as técnicas modelagem computacional para o estudo de sistemas nanoestruturados (constituído de partes que vão de dezenas a milhares de átomos) e também de sistemas macroscópicos. Além disso, o grupo se dedica ao desenvolvimento de novas metodologias, como, por exemplo, para o estudo de eventos raros ou o tratamento de várias escalas de tamanho. São basicamente quatro linhas de pesquisa:
Uma metodologia diversificada é utilizada nesses estudos. A modelagem dos sistemas físicos inclui desde os sofisticados métodos ab initio (que partem de descrições quânticas detalhadas dos átomos e moléculas, incluindo seus elétrons), baseados na Teoria do Funcional de Densidade (DFT), passando por métodos híbridos como a aproximação tight-binding até a técnica dos potenciais efetivos, que podem variar desde potenciais bastante sofisticados (analíticos ou numéricos) até potencias mais simples como o de Lennard-Jones. Nessa modelagem, os sistemas físicos são simulados através de métodos como o Monte Carlo e a Dinâmica Molecular.
Os estudos do grupo têm por objetivo entender fenômenos ainda não bem compreendidos, como as transições vítreas e líquido-líquido e os defeitos de diversos tipos em cristais, como também o aperfeiçoamento dos métodos computacionais. Neste último caso, os objetivos são três: (1) descrever os sistemas de forma mais precisa; (2) conseguir fazer simulações em escalas de tempo cada vez maiores e (3) englobar escalas de tamanho cada vez maiores (o maior número possível de partículas).
Dependendo do modelo utilizado, o tamanho do sistema simulado pode ir de dezenas a alguns milhares de átomos. Para potenciais simples, como o de Lennard-Jones, pode-se chegar a um bilhão de átomos ou mais. Métodos mais detalhados, como o DFT, exigem redução drástica desse tamanho. A paralelização dos códigos computacionais tem permitido fazer simulações de sistemas cada vez maiores. Entretanto, no que diz respeito à simulações em tempos muito longos, esforços metodológicos ainda são necessários para que as escalas de tempo de simulação se aproximem das escalas de tempo experimentais.
São sistemas constituídos por partes nanométricas, isto é, com nanômetros de diâmetro. Podem ter diversas formas – nanofios, nanotubos, nanoclusters (que são pequenos aglomerados de dezenas de átomos; o Grupo estudou, por exemplo, como nanoclusters depositados sobre uma superfície difundem-se espontaneamente pelo material adentro) e filmes finos (poucas camadas atômicas) depositados em superfícies metálicas.
Uma porção de certo material com tamanho tão pequeno possui propriedades diferentes de quantidades macroscópicas do mesmo material, pois as propriedades de sua superfície passam a interferir de modo preponderante nas suas características como um todo (as propriedades da superfície podem ser bastante diferentes das do resto do volume da substância). Por isso, esses sistemas possuem inúmeras propriedades novas que estão ainda sendo investigadas, tanto experimentalmente quanto por simulação computacional (que é o caso do Grupo). É a nova área da nanociência e da nanotecnologia.
No estudo de nanofios metálicos, o Grupo estudou com sucesso a formação e quebra de um fio muito fino de ouro que, antes de quebrar, forma uma cadeia linear com apenas alguns átomos de comprimento. O primeiro trabalho desta linha de pesquisa foi matéria de capa da revista Physical Review Letters (PRL) (figura abaixo, à esquerda) e, devido a esse destaque, foi também matéria de capa da revista Pesquisa Fapesp (figura abaixo, à direita). Trabalho posterior, também publicado no PRL, foi destacado pelo editor da revista Science, na seção “Editors Choice”, no artigo “A Bit of Stretch”.

Fig 1 - Capa da revista Physical Review Letters; o trabalho do Grupo em nanofios de ouro foi o destaque daquele fascículo
(Silva et al., Phys. Rev. Lett. 87, 256102 (2001))

Fig 2 - Capa da revista Pesquisa Fapesp,destacando a pesquisa em nanofios de ouro e o trabalho do Grupo na PRL
(Silva et al., Phys. Rev. Lett. 87, 256102 (2001))
Além do intenso trabalho em nanofios com espessura de um átomo, que têm sido estudados em estruturas de ouro e de cobre, estudamos também fios metálicos com diâmetros de alguns nanômetros, nos quais é possível um novo e importante processo, a solda a frio (cold welding). Isto é, solda sem o uso de calor, metais derretidos ou altas pressões. A equipe mostrou, em simulações computacionais, que este processo ocorre com resultados surpreendentes.
Fases são maneiras com que a matérias se organiza a uma dada condição de pressão, temperatura etc. Por exemplo, sólido, líquido e gasoso – a passagem de um para o outro é uma transição de fase. O grupo estuda transições de fase de primeira ordem, que são as descontínuas (quando o líquido passa para gasoso, diversas propriedades, como volume e densidade, mudam abruptamente). Nas transições de segunda ordem, não há variação abrupta das grandezas físicas. As transições de primeira ordem também se caracterizam por acontecerem primeiro em pequenas regiões do material para depois se estenderem para todo ele – o chamado processo de nucleação.
O Grupo investiga especialmente transições de fase em ligas metálicas, semicondutores e metais, por meio da análise de como as variáveis termodinâmicas mudam durante a transição. A principal variável investigada é a chamada energia livre, pois com ela pode-se obter todas as propriedades termodinâmicas relevantes de um sistema: pressão, volume, entropia etc.
Os cristais caracterizam-se por terem seus átomos ou moléculas organizados num ordenamento periódico que se repete por todo o material. Isso, pelo menos, é o que seria um cristal perfeito, mas, na prática, eles apresentam pequenos defeitos. Dependendo do caso, esses defeitos, mesmo tendo tamanhos em escala atômica e sendo em minúscula quantidade, podem ter influências determinantes nas características do material como um todo. Como isso acontece é o que o Grupo investiga.
Exemplos de defeitos são a vacância (ausência de um átomo em certa posição no cristal) e o interstício (presença de um átomo a mais no meio da rede cristalina). Esses defeitos podem deslocar-se espontaneamente dentro do material e difundir-se pelo seu interior.
Outro tipo importante de defeito é o defeito topológico, que não consiste em partículas faltantes ou a mais, mas em outras formas de organização das mesmas. Um exemplo: no caso do gelo, as moléculas de água formam uma estrutura como na figura ao lado. Note que a estrutura é essencialmente definida pela posição dos átomos de oxigênio da molécula. Os de hidrogênio, porém, podem variar (a molécula pode estar com diferentes orientações). Essa variação, quando ocorre, é que constitui o defeito topológico, no caso descrito no parágrafo anterior.

Estrutura do gelo. Os átomos de oxigênio estão representados em vermelho e os de hidrogênio, com pequenas bolinhas brancas. As linhas tracejadas são ligações (pontes de hidrogênio) entre as moléculas de H2O
Há, então, vários tipos de gelo, dependendo do modo como as moléculas de água se orientam e de outros tipos de parâmetro. O gelo possui nada menos que 13 fases sólidas distintas. Esses defeitos causam alterações em certas características globais do gelo, como sua condutividade elétrica. Até hoje esses defeitos topológicos não são completamente compreendidos.
A expressão “transição líquido-líquido” pode parecer estranha para quem está acostumado às transições de fase sólido-líquido, líquido-gasoso etc. Acontece que os líquidos, apesar de suas moléculas estarem em constante movimento e mudarem de forma todo o tempo, possuem uma estrutura em pequena escala, constituída pelo modo como cada molécula liga com suas vizinhas. Assim, no silício sólido, cada átomo possui 4 vizinhos; no líquido, porém, cada um possui 6 vizinhos. Outra diferença é que, no caso sólido, os 4 vizinhos são sempre os mesmos, estão fixos; no caso líquido, os átomos trocam de posição, entram e saem da vizinhança dos outros átomos o tempo todo, mas o padrão de 6 vizinhos para cada um se mantém, dinamicamente.
Mudanças nessa estrutura mantendo o estado líquido são um exemplo do que se entende por transição de fase líquido-líquido. Macroscopicamente, essas transições alteram as propriedades do material, como sua densidade e sua viscosidade. É um fenômeno muito difícil de ser estudado experimentalmente; por isso, há vários casos ao redor do mundo de transições líquido-líquido encontradas em simulações computacionais mas ainda sem confirmação experimental. Os dois únicos casos em que houve confirmação inequívoca são o fósforo e um tipo de óxido misto de alumínio e ítrio. O Grupo, por exemplo, já demonstrou a existência de uma transição líquido-líquido no gálio puro por meio de simulações computacionais.
Outro tipo de transição investigada pelo Grupo é a vítrea. Normalmente, um líquido, quando se congela, torna-se um cristal, seus átomos e moléculas passam a ordenar-se numa rede cristalina por todo o material. Algumas substâncias, porém, não se cristalizam. Chama-se sólido amorfo ao sólido que não é cristalino. O vidro – óxido de silício – é um deles; por isso, o fenômeno é chamado transição vítrea. São sistemas que despertam muito interesse por terem características semelhantes a alguns sistemas de interesse biológico, como proteínas.
Trata-se de um fenômeno ainda não bem compreendido. Uma hipótese é que existam vários mínimos de energia próximos do congelamento, cada um associado a uma configuração (cristalino e diversas formas de não-cristalino); se a temperatura abaixar muito rapidamente, o material fica preso num desses mínimos, associado à fase amorfa, e “não encontra” o mínimo principal correspondente à fase líquida (nem todo amorfo é vítreo, porém; os vítreos são obtidos por resfriamento e os amorfos não-vítreos, por outros métodos).
Nessas áreas, o Grupo investiga em particular a dinâmica da transição líquido-líquido – como ela acontece. Algumas substâncias, por exemplo, passam por uma fase intermediária, na qual ficam mais viscosas. Novamente, é um fenômeno pouco compreendido, especialmente certos materiais que ficam menos densos ao mesmo tempo em que sua viscosidade aumenta – um fenômeno totalmente contra a intuição. Isso acontece com o gálio. Especula-se que, na fase mais viscosa, seus átomos se associem em pequenos aglomerados com espaços vazios no meio (como “gaiolas”), o que faz com que sua densidade diminua ao mesmo tempo que dificulta a sua viscosidade.
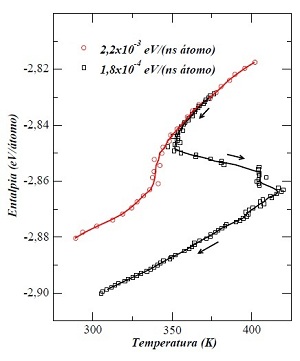
Este gráfico é um exemplo de como se pode detectar teoricamente a existência de transições líquido-líquido em um material - no caso, o gálio. Os cálculos que originaram o gráfico simulam um experimento no qual se retira energia do gálio lentamente. A curva vermelha é para uma taxa de retirada de energia maior, a preta, para uma menor. Com o correr do tempo, o sistema vai percorrendo as curvas de cima para baixo. A temperatura vai diminuindo com o tempo, mas é visível a mudança de comportamento da mesma quando ela alcança perto de 350 kelvins. Para a curva preta, chega a haver uma reversão na diminuição da temperatura, que passa a aumentar, para depois voltar a diminuir. Tais mudanças de comportamento são típicas de transições de fase. No caso, indicam uma transição líquido-líquido
Fonte: Tese de doutorado de Mateus Fontana Michelon, IFGW/Unicamp (2009), pág. 54
A equipe possui um sistema computacional SGI adquirido usando verbas de vários projetos, FAPESP, CNPQ e Capes. Neste momento, o computador tem as características apresentadas abaixo.
Este sistema receberá um upgrade em 2011, que deve duplicar sua capacidade computacional.

Alem deste sistema, os pesquisadores e alunos do grupo também utilizam recursos do parque computacional do IFGW e também do parque computacional do Centro Nacional de Processamento de Alto Desempenho em São Paulo (CENAPAD-SP).
Este grupo de pesquisas iniciou suas atividades na década de 1980 com estudos de estrutura eletrônica de metais e ligas metálicas. Na década de 90 passou também a estudar estrutura eletrônica de semicondutores. Nessa década se iniciaram os estudos de propriedades termodinâmicas usando técnicas de dinâmica molecular ab initio, dinâmica molecular com potenciais efetivos e Método de Monte Carlo. Através dessas novas técnicas iniciaram-se os estudos de transições de fase. Na primeira década do novo século iniciaram as pesquisas em nanociência com estudos de nanofios metálicos nanotubos de carbono e em grafeno.
Universidade Estadual de Campinas - Instituto de Física Gleb Wataghin
Rua Sérgio Buarque de Holanda, 777
Cidade Universitária, Campinas - SP, 13083-859
Fone +55 19 3521-5297
Fax +55 19 3521-4147